
Hu Translational Lab

About the Lab:
The translational laboratory at the Department of Emergency Medicine UCSD has a wealth of research experience spanning more than 25 years. The lab has specialized in utilizing animal models of ischemic stroke, hemorrhagic shock, cardiac arrest, neonatal hypoxia-ischemia, and traumatic brain injury to investigate intracellular membrane trafficking, the endolysosomal cycle, organelle-specific autophagy, protein misfolding and aggregation, and the effects and benefits of focal therapeutic hypothermia. We employ multidisciplinary and state-of-the-art genetically modified animal models, advanced tissue staining and microscopy techniques, molecular biological techniques, and MRI imaging to enhance our understanding of molecular mechanisms and the effects of therapeutic interventions on tissue ischemia-reperfusion injury. Our research team comprises professionals specializing in animal surgery, behavioral monitoring and analysis, molecular biology, therapeutic interventions, and MRI imaging in experimental, preclinical, and clinical research.
Research Interests:
Stroke, cardiac arrest, hemorrhagic shock, and traumatic injuries are among the major life-threatening conditions seen in the emergency department. Patients suffering from these medical emergencies requires timely diagnosis and treatment in the emergency room to prevent short-term mortality as well as long-term disabilities. The common features of these emergency conditions are ischemia-reperfusion injury, neurodegeneration, as well as local and systemic inflammation. As an emergency medicine translational research laboratory, our interests are focused on understanding the molecular mechanisms of tissue damage in these pathological conditions. We aim to develop brain protective interventions to prevent or stop the progression to irreversible brain injury and improve patient outcomes.
NIH SPAN program:
The NIH Stroke Pre-Clinical Assessment Network (SPAN) seeks to conduct late-stage preclinical studies of putative neuroprotectants combined with reperfusion. SPAN utilizes a novel, adaptive, secured system for parallel testing of promising interventions designed to extend the treatment time window and/or improve outcome compared to reperfusion when combined with thrombolysis, thrombectomy or both. SPAN was established to address a significant need in the scientific investigation of stroke treatment. In the past, a plethora of putative neuroprotectants proceeded to clinical trial based on favorable preclinical assessment, only to fail in subsequent clinical trials of human stroke patients. The recent successful development of thrombectomy for acute ischemic stroke generated considerable enthusiasm for re-testing treatment candidates in combination with thrombectomy. Thus, SPAN is intended to screen and select highly promising treatment candidates for possible further study in human clinical trials. SPAN has six testing sites/centers around the United States including the UCSD SPAN lab.
For more information, please see: https://spannetwork.org.

Current Projects and Support:
- NIH U01 4/1/2022 – 3/31/2026 entitled: Testing Cerebroprotective Interventions with Rodent Ischemic Stroke Models. Major Goals: To test cerebroprotective interventions for the SPAN program.
- NIH R01 7/1/2023 – 6/30/2028 entitled: The Role of Lysosomal Membrane Permeabilization and Cathespin B Release in Stroke Brain Injury. Major Goals: To study the role of endolysosomal structural damage and release of cathepsin B in brain ischemia reperfusion injury.
- VA Merit Award 07/01/2023 - 06/30/2027 entitled: Novel Anti-Stroke Agents Targeting Toxic Protein Aggregation. Major Goals: This proposal aims to investigate the novel agents that can reduce protein misfolding and aggregation in post-ischemic neurons using a mouse model of focal brain ischemia.
- NIH R01 01/01/2018 – 12/31/2024 (NCE) entitled: Change in NSF ATPase activity Leads to Brain Ischemia Reperfusion Injury. Major Goals: The major goals of this project are to understand the mechanisms and consequences of damage to the intra-neuronal Golgi apparatus, Golgi-derived transporting vesicles (Vs) and late endosomes (LEs) (i.e., Golgi/V/LE damage) after brain ischemia.
- NIH R01 04/01/2017 – 06/30/2024 (NCE) entitled: Novel anti-NPC aggregation strategy against brain ischemia-reperfusion injury. Major Goals: The major goal of this study is to develop a new therapeutic strategy for reducing the load of the newly synthesized polypeptides using mouse models of stroke.
- DOD Award 08/31/2018 - 09/15/2023 (NCE) entitled: Novel Focal Abdominal Cavity Cooling REBOA Strategy for the Treatment of Noncompressible Torso Hemorrhage. Major Goals: This project is to study focal cooling after hemorrhagic shock.
Example Project
One of the current working hypotheses in the lab is that the inactivation of a key membrane trafficking protein called N-ethylmaleimide sensitive factor (NSF) leads to release of damaging hydrolases, contributing to ischemic stroke brain injury.
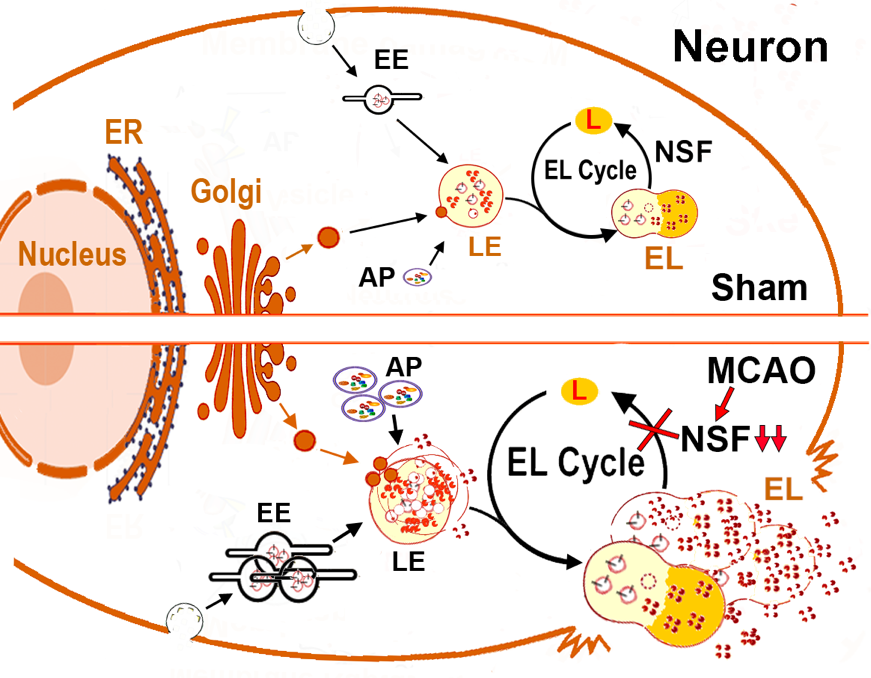
Figure Legend: Schematic Diagram of Post-Stroke Release of lysosomal hydrolases. Upper: In sham neurons, lysosomal hydrolytic enzymes and lysosomal structural proteins are synthesized on the ER-associated polyribosomes, modified in the ER and the Golgi lumen, and transported via vesicles to the late endosome (LE). The LE also receives incoming waste cargos from early endosome (EE) of the endocytic pathway and autophagosome (AP) of the autophagic pathway. The enzyme- and waste cargo-loaded LE then enters the lysosomal degradation cycle (L-Cycle) by fusing with an acidic terminal lysosome (L) for the formation of an autolysosome (AL) or endolysosome (EL) where the cargo can be degraded in an acidic environment (~pH 4.5). After degradation of waste cargo, AL or EL converts into a new L via an NSF-mediated mechanism for the next round of LE-to-L fusion. Lower: stroke or brain ischemia leads to NSF deficiency in neurons, thus causing disruption of the AL’s/EL’s conversion to a new L. This results in the endocytic and autophagic traffic jams. Consequently, both the size and number of all the structures related to the endocytic and autophagic pathways before the AL/EL-to-L conversion phase, are increased. This cascade of events eventually damages the lysosome-related structures to release hydrolases into the cytoplasm or extracellular space contributing to ischemic stroke brain injury.
Highlighted Publications:
- Arya AK, Hu K, Chen A, Olivas-Garcia Y, Coyne C, Tanaka H, Liu C, Doucet J, Chan T, Hu B. Intracolon cooling increases survival rate in the rat model of lethal hemorrhage. Shock. 2023 Dec 1;60(6):762-770.
- Hu K, Park Y, Olivas Y, Chen A, Liu C, Hu B. Cathepsin B knockout confers significant brain protection in the mouse model of stroke. Exp Neurol. 2023 Oct;368:114499.
- Yuan D, Hu K, Loke CM, Teramoto H, Liu C, Hu B. Interruption of endolysosomal trafficking leads to stroke brain injury. Exp Neurol. 2021;345:113827.
- Yuan D, Liu C, Wu J, Hu B. Nest-building activity as a reproducible and long-term stroke deficit test in a mouse model of stroke. Brain Behav. 2018;8(6):e00993.
- Yuan D, Liu C, Hu B. Dysfunction of Membrane Trafficking Leads to Ischemia-Reperfusion Injury After Transient Cerebral Ischemia. Transl Stroke Res. 2018;9(3):215-22.
- Liu C, Gao Y, Barrett J, Hu B. Autophagy and protein aggregation after brain ischemia. J Neurochem. 2010;115(1):68-78.
- Ge P, Luo Y, Liu CL, Hu B. Protein aggregation and proteasome dysfunction after brain ischemia. Stroke. 2007;38(12):3230-6.
- Zhang F, Liu CL, Hu BR. Irreversible aggregation of protein synthesis machinery after focal brain ischemia. J Neurochem. 2006;98(1):102-12.
- Liu CL, Ge P, Zhang F, Hu BR. Co-translational protein aggregation after transient cerebral ischemia. Neuroscience. 2005;134(4):1273-84.
- Hu BR, Janelidze S, Ginsberg MD, Busto R, Perez-Pinzon M, Sick TJ, et al. Protein aggregation after focal brain ischemia and reperfusion. J Cereb Blood Flow Metab. 2001;21(7):865-75.
- Hu BR, Janelidze S, Ginsberg MD, Busto R, Perez-Pinzon M, Sick TJ, et al. Protein aggregation after focal brain ischemia and reperfusion. J Cereb Blood Flow Metab. 2001;21(7):865-75.
- Hu BR, Martone ME, Jones YZ, Liu CL. Protein aggregation after transient cerebral ischemia. J Neurosci. 2000;20(9):3191-9.